загальність
Синдром Прадера-Віллі - рідкісне генетичне захворювання, яке викликає фізичні, поведінкові та інтелектуальні порушення. Найбільш характерними клінічними ознаками є ожиріння (і супутні захворювання) і зниження м'язового тонусу.
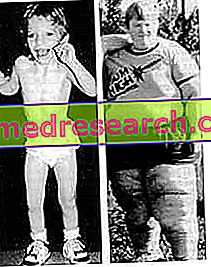
Фізичне обстеження, як правило, достатньо для встановлення правильного діагнозу, але також можна провести достовірні генетичні тести.
На жаль, досі немає остаточної терапії; однак деякі фармакологічні та поведінкові контрзаходи можуть обмежувати пов'язану симптоматику.
Короткий нагадування про генетику
Перш ніж описувати синдром Прадера-Віллі, добре зробити коротку посилання на генетику.
ХРОМОСОМИ І ДНК
Кожна клітина здорової людини має 23 пари гомологічних хромосом : 23 - материнські, тобто успадковані від матері, і 23 - батьківські, або успадковані від батька. Пара цих хромосом статева, тобто вона визначає стать індивідуума; решта 22 пари, замість цього, складаються з аутосомних хромосом . В цілому, 46 людських хромосом містять весь генетичний матеріал, більш відомий як ДНК . У ДНК індивіда написані його соматичні риси, його схильність, його фізичні здібності і т.д.
ГЕНИ І МУТАЦІЇ ДНК
ДНК організована в безлічі послідовностей, більш-менш довгих, званих генами .

Малюнок: організація гена в межах пари гомологічних хромосом. Пара гомологічних хромосом містить специфічні гени, всі з двома варіантами, алелі, що займають одне і те ж хромосомне положення і виконують ті ж функції (крім мутацій). Ліва пара хромосом має два рівних алеля (обидва синіх); Права пара має два різних алеля (один червоний, другий - синій).
Кожен ген займає певну хромосому і її аналог, оскільки він присутній у двох копіях, званих алелями . Алель походить від матері і знаходиться в материнській хромосомі; інший алель походить від батька і знаходиться в батьківській хромосомі.
З генів відбуваються білки, присутні в нашому тілі. Коли відбувається мутація ДНК, ген (зазвичай алель) даної хромосоми може бути дефектним і тому виробляти дефектний білок або взагалі відсутній.
Що таке синдром Прадера-Віллі?
Синдром Прадера-Віллі - рідкісне генетичне захворювання, яке характеризується низкою фізичних, інтелектуальних та поведінкових дефіцитів, що залежать від зміни хромосоми 15.
У ранньому дитинстві синдром проявляється незвичайною м'язовою слабкістю і затримкою розвитку. Пізніше, в дитинстві, починають виникати інші проблеми, такі як постійний апетит, труднощі в навчанні та поведінкові аномалії.
Особи, що мають синдром Прадера-Віллі, дуже часто є людьми, які страждають ожирінням, від яких виникають різні серцеві проблеми. Це головна причина смерті.
епідеміологія
Синдром Прадера-Віллі - рідкісне захворювання: насправді народження ураженої дитини реєструється в середньому кожні 15 000–30 000 новонароджених.
Вона уражує чоловіків і жінок рівною мірою і не має пристрасті до певних порід.
причини
Причиною, що викликає синдром Прадера-Віллі, є генетична мутація на хромосомі 15 . Точний ген, що вразився, ще не з'ясований; підозри падають, більш ніж будь-що інше, на хромосомну область, яка включає більше генів.

Малюнок: хромосома 15 і гени, які підозрюються у ролі синдрому Прадера-Віллі. З сайту: www.kreatech.com
ГЕНИ НА ХРОМОСОМІ 15
Зазвичай клітини нашого організму використовують обидва алелі для створення білків. Іншими словами, це означає, що обидві хромосоми, материнські та батьківські, корисні і забезпечують свій генетичний внесок.
Однак у деяких окремих клітинах, внаслідок еволюційного та непатологічного питання, працює тільки один (батьківський або материнський) алель, і його робота більш ніж задовільна. Неактивний алель називається тихим, саме тому, що він існує, але він не «виражає» себе.
Клітини нашого мозку містять всі хромосоми 15, але в деяких регіонах виражена лише материнська генетична лінія, в інших - лише батьківська. У гіпоталамусі, який є ділянкою головного мозку, відповідальним за синдром Прадера-Віллі, зазвичай експресуються тільки гени батьківської хромосоми.
ХРОМОСОМА 15 І СИНДРОМ ПРАДЕР-ВІЛЛІ
За допомогою генетичного тестування було виявлено, що у пацієнтів з синдромом Прадера-Віллі бракує нормальної 15-ої хромосоми. Це є шкідливим для гіпоталамуса, де єдина активна хромосомна лінія є батьківською.
Основні функції гіпоталамуса:
- Регулювання апетиту
- Регулювання ритмів сну-сон
- Вираз емоційних станів
- Регулювання температури тіла
- Виробництво гормонів
Але що викликає несправність або відсутність 15-ої хромосоми батька? Є принаймні три можливі причини:
- Відсутність певної ділянки 15 батьківської хромосоми: насправді хромосома не має істотної частини.
- Дві хромосоми 15 материнські. Ця аномалія виникає через помилку при утворенні ембріона.
- Зміна деяких генів, присутніх на хромосомі батьківства 15.
ГЕНЕТИЧНИЙ ТА ГЕРЕДИТАРНИЙ? АБО ТІЛЬКИ ГЕНЕТИКИ?
Генетики вважають синдром Прадера-Віллі генетичним захворюванням, оскільки, згідно з дослідженнями, з'ясувалося, що три вищезазначені режими мутації не успадковані від батьків (які мають нормальне хромосомне спорядження), але виникають випадково., безпосередньо перед зачаттям ( спорадична мутація ).
Однак достовірність цього твердження підривається виявленням деяких пар батьків з більш ніж однією дитиною, яка страждає від синдрому Прадера-Віллі. У цих випадках є підстави вважати, що може виникнути деяка спадкова складова, яка ще доведеться продемонструвати, у походження хвороби.
СИНДРОМ ПРАДЕР-ВІЛЛІ І СИНДРОМ АНГЕЛЬМАНА
Синдром Прадера-Віллі є, у певному сенсі, протилежним синдрому Ангельмана : у останньому, насправді, саме хромосома 15 материнської людини не функціонує належним чином.
Симптоми та ускладнення
Дізнатися більше: Симптоми синдрому Прадера-Віллі
Синдром Прадера-Віллі проявляється, з першими симптомами і ознаками, вже в ранньому дитинстві (перший рік життя); в цей період він, головним чином, викликає зниження м'язового тонусу (гіпотонії) і затримку розвитку. Згодом хвороба переживає своєрідну еволюцію, яка додатково збагачує симптоматичну картину.
ДИТИНСТВО
Основними ознаками протягом першого року життя є:
- М'язова гіпотонія . Це означає, що тонус м'язів знижується в порівнянні з нормальним: зазвичай виявляється м'якими і не дуже реактивними кінцівками, а також важким ссанням молока матері.
- Затримка розвитку . Вона має тенденцію до сприятливих труднощів смоктання через гіпотонію.
- Косоокість .
- Характерні риси обличчя . Мигдалеподібні очі, звуження голови на скронях, рот вниз і тонка верхня губа.
- Часткова або повна відсутність реакції на подразники . Дитина втомилася і нелегко розбудити його.
З ВІКУ ДО ВІКУ? ДОРОСЛИХ
Починаючи з першого року життя, виникла довга серія проблем, які можуть мати драматичні результати.
- Чудовий апетит і ожиріння . Пацієнти демонструють постійне прагнення до їжі, що змушує їх багато їсти і отримувати значну вагу. Якщо їм нічого не їсти, вони приходять, щоб споживати заморожені продукти і відходи, іншими словами, щось їстівне. Все це обумовлено зміненими функціями гіпоталамуса.
- Гіпогонадизм . Це означає, що статеві органи (яєчка, у людей і яєчники, у жінок) виробляють кілька статевих гормонів (чоловічий тестостерон і жіночий естроген). Пацієнт не закінчує пубертатний розвиток і зазвичай не родючий. Перша менструація у жінок затримується (якщо не повністю відсутня); у людини не спостерігається ніякої зміни голосу.
- Зниження росту та розвитку . До проблеми м`язової гіпотонії, яка залишається, додається скорочений розвиток статуру навіть після періоду статевого дозрівання (в якому, як правило, підлітки піднімаються на кілька сантиметрів).
- Вивчення дефіциту . Інтелектуальні здібності пацієнтів майже завжди знижуються.
- Поведінкові проблеми . Особливо в підлітковому віці особини є впертими, примхливими і страждають від так званого обсесивно-компульсивного розладу.
- Затримка двигуна . Діти вчаться ходити дуже пізно.
- Мовні труднощі . Зазвичай пацієнти починають розмовляти зі значною затримкою, і їхня мова завжди залишається бідною і важкою.
- Розлади сну . Нормальне чергування між фазами REM і NON-REM не дотримується. Крім того, пацієнти, коли вони сплять, страждають від переривань дихання (апное сну).
- Сколіоз . Проблема стосується лише деяких пацієнтів.
КОЛИ ДЛЯ ВІДПОВІДАЛІ ДО ЛІКУ
У немовляті. Ознаками, які повинні спонукати вас підозрювати синдром Прадера-Віллі, є: відсутність розвитку, м'язова гіпотонія, труднощі з засмоктуванням грудного молока, особливості обличчя та відсутність реакції на подразники.
У дитини. Двома фундаментальними підказками є: наполегливий пошук продуктів і швидке збільшення ваги.
Ускладнення
Основні ускладнення синдрому Прадера-Віллі пов'язані з ожирінням і всіма супутніми проблемами, такими як цукровий діабет, серцеві захворювання, гіпертонія, гіперхолестеринемія, атеросклероз тощо. Більш того, залишаючись в контексті безперервного харчування, пацієнту легко задихатися через їжу, спожиту ненажерливо.
Інша серія дуже важливих ускладнень пов'язана з гіпогонадизмом : відсутність статевих гормонів дуже часто викликає стерильність і остеопороз .
діагностика
Перш ніж вдатися до генетичних тестів, можна провести правильну попередню діагностику синдрому Прадера-Віллі за допомогою простого фізичного обстеження та кількох аналізів крові.
Клінічні ознаки можуть бути виявлені при медичному огляді
у дитини:
- М'язова гіпотонія
- Мигдальні очі
- Скорочення храмів
У дитини / підлітка:
- Ненаситний апетит
- ожиріння
- Поведінкові проблеми
Генетичні тести служать підтвердженням і допомагають уточнити тип мутації, що викликав захворювання.
лікування
На жаль, оскільки це генетичне захворювання, синдром Прадера-Віллі не виліковний.
Єдиним застосовним терапевтичним методом лікування є обмеження симптомів (наприклад, ожиріння), пом'якшення деяких ненормальних форм поведінки і, як правило, поліпшення рівня життя пацієнтів.
Щоб досягти успіху в усьому цьому, доцільно звернутися до команди лікарів і фахівців, що спеціалізуються в різних областях - від ендокринології до дієтології, від фізіотерапії до психотерапії.
Найбільш поширені лікувальні заходи наведені нижче.
ХАРЧУВАННЯ В ДИТИНСТВІ ТА НАСТУПНИХ КРОКИ
У ранньому дитинстві, для подолання труднощів смоктання і відсутності розвитку, добре дати дитині висококалорійну їжу.
На наступних етапах ситуація повністю змінюється: прийом їжі повинен бути ретельно перевірений, приділяючи максимальну увагу калоріям.
Найбільш підходящим фахівцем для прохання про консультацію є дієтолог .
ГОРМОН РОСТУ
Екзогенне введення (тобто ззовні) гормону росту ( GH ) має три ефекти:
- Заохочення зростання, якого інакше бракувало б
- Поліпшення м'язового тонусу
- Знизити рівень жирової маси
Лікування починається у віці близько 3-5 років.
Сьогодні в лабораторії створені гормональні препарати, ефективні і з зменшеними побічними ефектами.
Найбільш зазначеним фахівцем, в даному випадку, є ендокринолог .
СЕКСУАЛЬНІ ГОРМОНИ
Екзогенне введення тестостерону для чоловіків і естрогенів для жінок має важливе значення для відновлення знижених рівнів цих двох гормонів. Крім поліпшення родючості, гормональна терапія має також вплив на остеопороз.
Лікування починається в період статевого дозрівання.
Дізнатися більше: Ліки для лікування синдрому Прадера-Вілі »
ФІЗІОТЕРАПІЯ І ЛОГОПЕДІЯ
Пацієнти з синдромом Прадера-Віллі потребують фізичної та мовної реабілітації . Перша спрямована на обмеження м'язової гіпотонії та наслідків ожиріння; другий засіб усунення недоліків спілкування, як розмовних, так і письмових.
Експертами, до яких звертатися, є, відповідно, фізіотерапевт і логопед.
ПСИХОТЕРАПІЯ І ТЕРАПІЯ ЗАЙНЯТОСТІ
Психотерапія необхідна для пацієнтів з обсесивно-компульсивними розладами і загалом настроєм. Підтримка психіатра або психолога може значно поліпшити поведінковий аспект.
Трудова терапія, з іншого боку, має на меті навчити пацієнта, як піклуватися про себе, як одягатися і т.д., іншими словами, як виконувати основні повсякденні заходи.
Допомога сімей
Близькість членів сім'ї необхідна для допомоги хворому родичу, особливо в молодості. Рекомендації, які зазвичай даються сім'ям, полягають у тому, щоб слідувати за пацієнтом у всіх його діях (особливо коли він годується), щоб довідатися про найбільш прийнятну поведінку, яку він може залишити для нього, не виключати його і т.д.
Прогноз і профілактика
Враховуючи, що синдром Прадера-Віллі є невиліковним захворюванням, прогноз ніколи не може бути позитивним. Найбільшу небезпеку представляють ожиріння і пов'язані з нею патології: смерть зазвичай обумовлена однією з них.
Доступні способи лікування (збалансоване харчування, гормональна терапія, психотерапія тощо) покращують якість життя, навіть у чутливому способі; однак, все ще існують заходи контролю симптомів і нічого більше.
Близькість родичів є фундаментальною: їх підтримка, по суті, може продовжити життя пацієнтів.
ПРОФІЛАКТИКА
Коли хвороба виникає в ембріона внаслідок генетичної мутації, її не можна запобігти.
Якщо замість цього два батьки вже народили дитину з синдромом Прадера-Віллі, то перед другою вагітністю вони можуть пройти специфічні генетичні тести, щоб з'ясувати, чи є вони носіями захворювання чи ні.


