Що таке синдром Марфана?
Синдром Марфана описує складний спадковий розлад сполучної тканини, який в основному вражає очі, серцево-судинну систему і систему скелетних м'язів. Однак, враховуючи, що кожен орган складається з сполучної тканини, синдром Марфана може ідеально зруйнувати і перешкоджати росту і функції кожного анатомічного сайту.
Синдром передається як аутосомно-домінантна ознака: тому ми стикаємося з серйозною генетичною хворобою, що має надзвичайно змінну фенотипову експресію (дефекти можуть значно відрізнятися від сім'ї до родини або від пацієнта до пацієнта).
Що викликає синдром Марфана - це зміна гена FBN1 (на хромосомі 15), який кодує фібриллін-1, дуже важливий сполучний глікопротеїн, який є структурною підтримкою мікрофібрил.
Мікрофібрили: що складаються з фибриллинов, микрофибрилли присутні в позаклітинному матриксі, в яких вони утворюють павутину для осадження еластину в еластичних волокнах. Незважаючи на всюдисущість в організмі, мікрофібрили рясніють в основному в аорті, зв'язках і зонах циліарних тіл (на рівні очей).
Будучи автосомно-домінантною хворобою, синдром Марфана впливають тільки діти, які успадкували гени FBN-1, змінені обома батьками. Проте, в одному з 4 випадків захворювання є результатом спонтанних мутацій у пацієнтів, які не мають сімейної історії.
Назва хвороби походить від французького педіатра, який вперше описав його в 1896 році (А. Марфан), після чого довелося чекати до 1991 року, щоб ідентифікувати змінений ген, що бере участь у симптоматичному прояві: першовідкривателем був Ф. Рамірес.
Перегляньте відео
X Перегляньте відео на YouTubeпричини
Ми згадали, що синдром Марфана є безпосереднім вираженням мутації гена, який кодує фібриллін-1. Спробуємо поглибити тему, щоб зрозуміти, який механізм створений для ініціювання синдрому.
FIBRILLINA 1 є глікопротеїновим компонентом еластину, необхідним для забезпечення та підтримки еластичності і міцності тканин. У фізіологічних умовах фібриллін 1 зв'язується з іншим білком, відомим як TGF-бета (або трансформуючим бета-фактором росту). Очевидно, що TGF-бета бере участь у шкідливих процесах, що впливають на гладкі м'язи судин і позаклітинний матрикс. Виходячи з цих припущень, деякі автори переконані, що синдром Марфана пов'язаний, крім мутації гена FBN-1, також з надлишком TGF-бета, особливо в аорті, в клапанах серця і в легенях.
падіння
Підраховано, що синдром Марфана впливає на 1 суб'єкта через кожні 3000-5000 народжень і виявляється нечітко між чоловіками і жінками, без пристрасті до точної породи. Статистика показує, що 75% пацієнтів мають позитивну сімейну історію; у решті 25% причина лежить в спорадичних мутаціях, які, здається, пов'язані, певним чином, з похилим віком батька на момент зачаття.
Діти з надзвичайно важкими формами синдрому Марфана мають тривалість життя менше одного року.
До еволюції хірургічних стратегій відкритого серця більшість пацієнтів з синдромом Марфана мали середню тривалість життя 32 роки; Завдяки постійному вдосконаленню медичних і фармакологічних терапій, в даний час хворі на синдром Марфана живуть в середньому до 60 років.
Ознаки та симптоми
Дізнатися більше: Симптоми синдрому Марфана
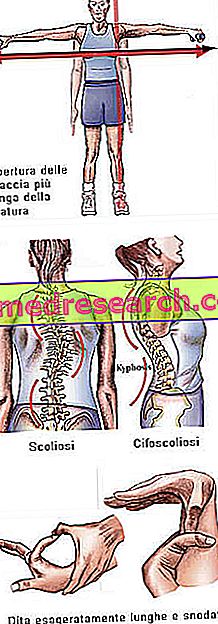
Синдром Марфана може протікати абсолютно безсимптомно. Уражені хворі мають надто струнку структуру, непропорційно високу і тонку. Нижні і верхні кінцівки мають набагато більшу довжину, ніж стовбур (долікостеномегалія). Існує також розмова про арахнодактилі для кращого вираження концепції перебільшеної довжини пальців, характерної для суб'єктів, що постраждали від синдрому Марфана: отже, руки порівнюються з ногами павука.
Що стосується висоти, то ці пацієнти мають середню висоту вище 97-го процентиля.
Інші відмінні риси, які часто зустрічаються у пацієнтів з синдромом Марфана:
- Руки відкриті більше, ніж висота
- В'ялі суглоби → перебільшена рухливість суглобів
- Деформація грудної стінки
- Кристалічне зміщення
- Верхня частина тіла менш розвинена, ніж нижня частина
- Спонтанний пневмоторакс (11%)
- сколіоз
- Шкірні смуги на стегнах, спині, дельтоподібному, грудному рівні
Серед найбільш проблемних ознак, пов'язаних з синдромом Марфана, ми згадуємо пролапс серцевого клапана і недостатність мітрального клапана: подібний стан може легко сприяти розширенню кільця аорти і розсічення аорти.
У таблиці наведено ознаки, які можна виявити у пацієнтів з синдромом Марфана. Описані там персонажі не завжди присутні, але більшу частину з них можна знайти.
| Уражений анатомічний сайт | Можливі симптоми |
милий | Стрий в грудному, поперековому і крижовому відділах |
очі | Погіршення зору, астигматизм, відшарування сітківки, вузькокутова глаукома, дислокація лінзи, короткозорість |
Структура кістки | Артралгія, кіфосколіоз, долікостенемія (надмірний розвиток у довжині кінцівки по відношенню до тулуба), гіпермобільність, високе піднебіння, деформовані груди, плоскі ноги, вузькі та тонкі зап'ястки, аномальне повернення / випинання грудини, сколіоз, вигнуті плечі, спондилолістез |
Dita | арахнодактилія |
легкі | Спонтанний пневмоторакс, задишка, ідіопатична легенева обструктивна хвороба |
Зміни обличчя | Огівальний піднебіння (пороки неба), нижня щелепа (дефект розвитку щелепи), витягнуте обличчя |
серце | Стенокардія, аневризма черевної аорти, серцева аритмія, розширення / розрив / розсікання грудної аорти, аортальна недостатність, пролапс мітрального клапана |
мова | Мова труднощі |
діагностика
Враховуючи більш ніж 200 можливих мутацій, використання генетичних маркерів практично неможливо для діагностичних цілей.
Встановлення синдрому Марфана не завжди настільки негайне, враховуючи, що фенотипова експресія мутації не завжди очевидна і проста для виявлення. Затримка діагностики може серйозно погіршити виживання пацієнта: просто подумайте, наприклад, про нездатність визнати серцево-судинну проблему.
Діагностичні критерії для синдрому Марфана були розроблені на міжнародному рівні в 1996 році: діагноз полягає в дослідженні сімейної історії, пов'язаної з поєднанням основних і другорядних показників синдрому.
Деякі з багатьох діагностичних тестів:
- ехокардіограма
- Магнітний ангіорезонанс і КТ (для дослідження аорти)
- магнітно-резонансна ангіографія (МРА) з контрастною рідиною (для виявлення внутрішніх структур аорти)
- огляд з щілинними лампами (для аналізу можливої дислокації об'єктива)
- вимірювання очного тиску (для висвітлення можливої присутності глаукоми)
- генетичні тести (рекомендується перед зачаттям дитини для встановлення синдрому чи ні)
терапії
Будучи генетичною хворобою, не існує ніякого препарату або лікування, яке може змінити хворобу.
Застосування препаратів, однак, є важливим для пом'якшення симптомів і уникнення будь-яких ускладнень, зокрема серцевих. Для цієї мети особливо придатні препарати для зниження артеріального тиску, такі як сартани (особливо), інгібітори АПФ і бета-блокатори.
У контексті синдрому Марфана пацієнти, які також страждають сколіозом, можуть слідувати специфічному догляду, а також тим, хто страждає глаукомою.
Можливо хірургічне втручання, щоб виправити аномальне розширення аорти, що часто об'єднує більшість пацієнтів з синдромом Марфана.
Продовжити: Синдром Марфана - наркотики та догляд »


